新藥查驗登記
- 更新日期:2025-02-08
- 點閱次數:13372
藥事法第三十九條第一項規定,製造、輸入藥品,應向中央衛生主管機關申請查驗登記,經核准發給藥品許可證後,始得製造或輸入。同法條第四項亦規定藥品查驗登記申請應依「藥品查驗登記審查準則」辦理。現行「藥品查驗登記審查準則」於九十四年一月七日公布,並經多次修正,該準則中規定各項藥品查驗登記應注意與遵循之事宜。另為有效運用審查資源以及增進民眾用藥可近性,中央衛生主管機關亦公告多項相關審查機制,以期使新藥加速上市,嘉惠病患。
以下內容將簡介如何申請新藥查驗登記,最新公告請至台灣藥物法規資訊網查詢。
- 查驗登記類別
- 藥事法第七條所稱之新藥:新成分、新使用途徑、新複方、新療效
- 其它:新劑型、新使用劑量、新單位含量製劑
- 生物藥品:基因工程藥品(含生物相似性藥品)、疫苗類藥品、人用血漿藥品、過敏原藥品、其他類
- 核醫放射性藥品
- 審查機制
新藥查驗登記除一般審查程序外,亦包括精簡審查、優先審查、加速核准、藥品突破性治療及小兒或少數嚴重疾病藥品審查等各種審查機制。
- 新藥查驗登記精簡審查機制:
詳見108年11月18日 衛授食字第1081410630號公告。適用對象(1~3)項皆須符合 - 第一類精簡審查:審查天數為180天
- 屬新成分新藥。
- 具有美國FDA、歐盟EMA或日本MHLW其中兩地區核准證明。
- 經評估未具族群差異者。
- 第二類精簡審查:審查天數為120天
- 屬新成分新藥。
- 有美國FDA、歐盟EMA 及日本MHLW核准證明且化學製造管制(CMC)資料皆相同。
- 經評估未具族群差異者。
所需文件 - 事先提出適用本機制之認定申請,並於查驗登記時檢附精簡審查認定同意函、查驗登記相關資料與規費。
- 美國FDA、歐盟EMA或日本MHLW/PMDA(獨立行政法人醫藥品醫療器材綜合機構)之核准證明、審查報告(Assessment report)及仿單。
- 針對美國FDA、歐盟EMA或日本MHLW/PMDA所要求之藥品風險管理計畫(Risk Management Plan;RMP)及上市後承諾(Post-marketing Commitment)提出最新進度報告。
- 免除銜接性試驗證明文件,且非有條件准予免除。
- 檢附資料應符合通用技術文件(CTD)格式且與檢送美國FDA、歐盟EMA或日本MHLW/PMDA相同。
說明 - 第一類精簡審查:審查天數為180天
- 新藥查驗登記優先審查機制:
詳見108年11月18日 衛授食字第1081410630號公告。適用對象(符合3項中之2項) - 屬「藥事法」第七條定義之新藥。
- 同時符合下列二條件:
- 適應症為我國的嚴重疾病。
- 滿足我國醫療迫切需求,係指具有醫療主要優勢(major advance)。
- 經我國政府核准優先輔導、補助研發,且具我國公共衛生或醫療迫切需求者。
所需文件 事先提出適用本機制之認定申請,並於查驗登記時檢附優先審查認定同意函、查驗登記相關資料與規費。 說明 - 審查天數為240天。
- 審查標準不因優先審查而改變,新藥應符合安全、療效與品質之要求,始准予上市。
- 新藥查驗登記加速核准機制:
詳見108年11月18日 衛授食字第1081410630號公告。適用對象(1~2)項皆須符合 - 屬「藥事法」第七條定義之新藥。
- 宣稱之適應症符合下列情形之一:
- 為我國的嚴重疾病,並能滿足我國醫療迫切需求(unmet medical need)。
- 具醫療迫切需求,且在十大醫藥先進國*之任一國已取得罕藥認定(Orphan drug designation)。
* 十大醫藥先進國為德國、美國、英國、法國、日本、瑞士、加拿大、澳洲、比利時、瑞典。 - 具醫療迫切需求,且於國內非屬罕見疾病藥物,製造或輸入我國確有困難者。
所需文件 事先提出申請適用本機制之認定,並於查驗登記時檢附加速核准認定同意函、查驗登記相關資料與規費。 說明 - 審查天數為240天。
- 在科學證據支持下,臨床試驗中得以替代療效指標做為間接或取代的評估方法,來代表具有臨床意義的改善成果,以縮短藥品研發上市時間。
- 經由本機制核准上市之藥品,原則上必須進行確認性試驗(confirmatory trials)以證明確實達到臨床上的效益。
- 申請人應於申請查驗登記時提出前述確認性試驗的試驗設計與完成試驗及報告時間。
- 藥品突破性治療認定要點:
詳見108年11月18日 衛授食字第1081410630號公告。適用對象(1~3)項皆須符合 - 屬藥事法之新成分新藥或已取得藥品許可證,且宣稱之適應症為我國嚴重疾病或罕見疾病。
- 早期臨床證據顯示其臨床療效指標比現行療法具重大突破性改善。
- 於我國執行有臨床意義之臨床試驗,如為早期臨床試驗尤佳。
所需文件 事先提出適用本機制之認定申請,並於查驗登記時檢附藥品突破性治療認定同意函、查驗登記相關資料與規費。 說明 - 雙向溝通機制:應於通過認定後至少每3個月向衛生福利部食品藥物管理署報告該案執行進度及產品未來研發規劃。其案件研發期間,如有待協助解決之法規相關議題,得向衛生福利部食品藥物管理署提出諮詢。
- 模組批次審查:得於申請查驗登記前,依據通用技術文件格式,向醫藥品查驗中心申請模組批次審查。
- 經認定適用本要點之藥品,於申請查驗登記時,宣稱之適應症與認定時相符者,得直接適用「新藥查驗登記優先審查機制」,審查天數為240天。
- 小兒或少數嚴重疾病藥品審查認定要點:
詳見108年11月18日 衛授食字第1081410630號公告。適用對象(1~4)項皆須符合 - 屬藥事法第七條定義之新藥。
- 適應症為我國的嚴重疾病。
- 該疾病主要影響小兒族群或盛行率在萬分之五以下。
- 滿足我國醫療迫切需求(unmet medical need)。
所需文件 事先提出適用本機制之認定申請,並於查驗登記時檢附小兒或少數嚴重疾病藥品審查認定同意函、查驗登記相關資料與規費。 說明 - 經本要點認定之藥品,以相同適應症申請藥品查驗登記時:
- 得直接適用「新藥查驗登記優先審查機制」,審查天數為240天。
- 無須事先提出銜接性試驗評估(BSE)申請,族群差異將於查驗登記案一併評估。
- 依藥品查驗登記審查準則,得免除採用證明,惟仍需於檢附出產國許可製售證明,倘該藥品於國內執行所申請適應症相關臨床試驗,得免除採用證明及出產國許可製售證明。
- 考量藥品為治療小兒或少數嚴重疾病,審查時臨床試驗之數目、受試者人數等方面得依個案適度放寬。
- 有關安定性試驗報告,原則上應檢送3批安定性試驗結果,若有特殊情況,得說明相關原因,先檢附1批安定性試驗結果送審。
- 經本要點認定為小兒嚴重疾病之新成分新藥,且其用以確認小兒族群療效、安全及用法用量之小兒族群臨床試驗於國內執行者,於該藥品核准取得許可證時,可取得優先審查憑證(voucher)。
- 新藥查驗登記精簡審查機制:
- 申請應檢附資料
- 申請案須一律附上「藥品管理類人民申請案_案件類別表」、「案件基本資料表」以便收文之順暢。
(請見100年9月19日署授食字第1001405584號公告及106年06月27日FDA藥字第1061405824號)。 - 藥品(不含學名藥及原料藥)查驗登記退件機制(Refuse to File;RTF)查檢表:(請見113年6月18日衛授食字第1131406955號公告),表內所提及之資料表及聲明表請詳見下述公告。
- 「GCP查核併藥品查驗登記申請案之臨床試驗資料表」(請見113年1月5日衛授食字第1121414566號公告)
- 「資料專屬期及國內外臨床試驗資料表」(請見113年6月18日衛授食字第1131406955號公告)
- 「藥品專利狀態之聲明表」(請見109年01月15日FDA藥字第1091400039號函)
- 各類別藥品申請查驗登記應檢附資料,請參考藥品查驗登記準則與相關附件:
- 申請案須一律附上「藥品管理類人民申請案_案件類別表」、「案件基本資料表」以便收文之順暢。
- 申請查驗登記時,得選擇下列任一方式送件:
- 紙本送件:依照衛生福利部食品藥物管理署公告之「CTD格式」,自行製作2份電子資料光碟送件,並以PDF文字檔為主。
- 線上送件:透過藥品查驗登記審查暨線上申請作業平台(ExPress)線上送案或是eCTD送件。
備註:使用eCTD送件應符合,請見FDA藥字第1101411462號函之相關規定
- 收件窗口:
| 案件狀態 | 受文者 | 紙本收件地址 | 線上送件 |
|---|---|---|---|
| 新申請案/申復案 | 衛生福利部食品藥物管理署 |
國家生技園區F棟(地址:11571台北市南港區研究院路一段130 |
藥品查驗登記審查暨線上申請作業平臺(ExPRESS) (請見衛生福利部食品藥物管理署網站>業務專區>藥品>藥品查驗登記線上申請) |
| 補件/申請補件延期 | 正本:財團法人醫藥品查驗中心 | 財團法人醫藥品查驗中心 (地址:11557台北市南港區忠孝東路六段465號3樓) |
|
| 副本:衛生福利部食品藥物管理署 | 國家生技園區F棟 (地址:11571台北市南港區研究院路一段130巷99號) |
三、審查流程
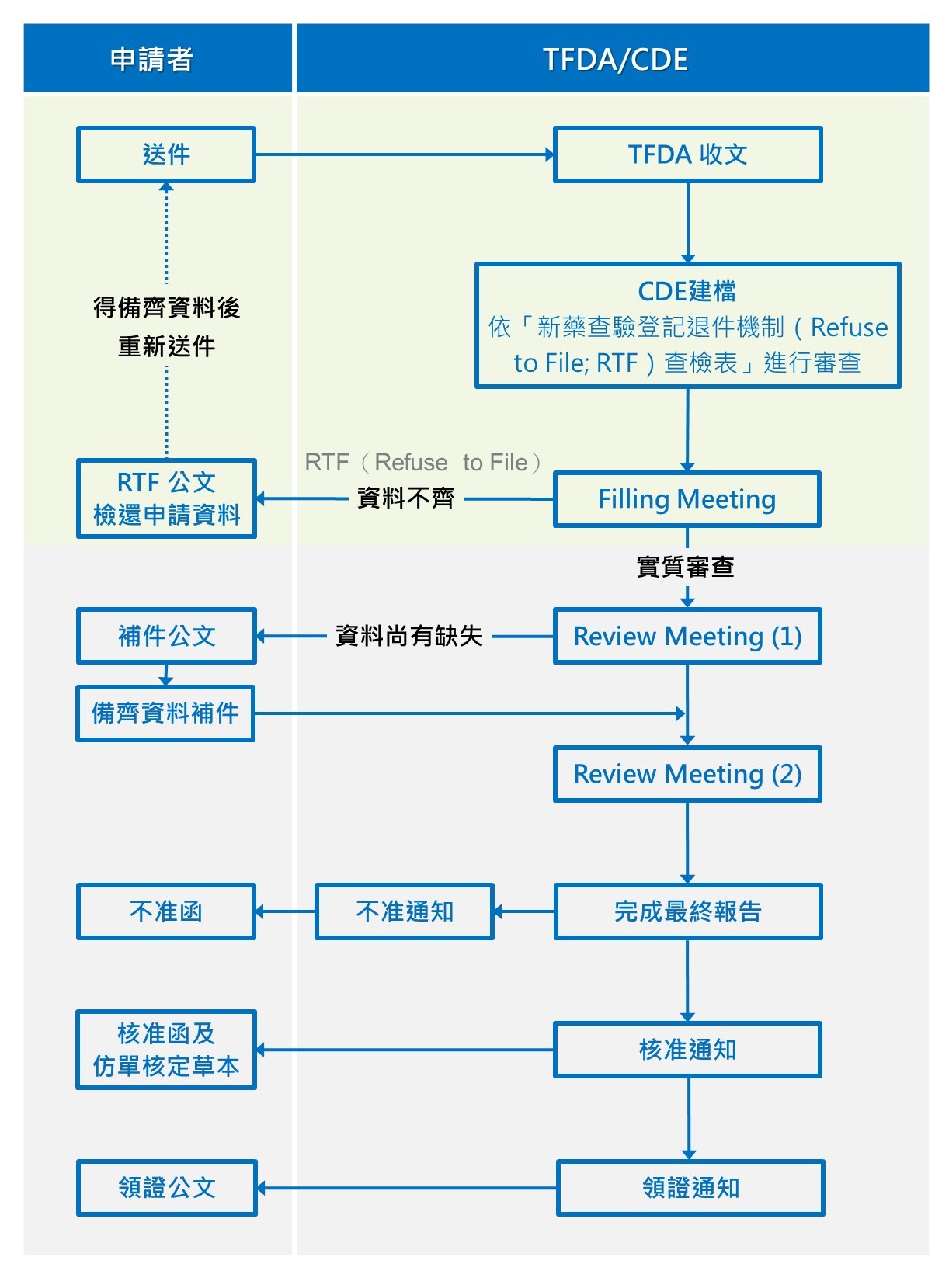
- 相關注意事項:
- 申請人如未能於期限內補正者,得於補正期滿前,以書面敘明理由申請延期;其延期期限,自補正期滿翌日起算一個月,且延期以一次為限。
- 基於風險管理原則,技術性資料審查後,如有特殊議題須諮詢專家委員意見,將視需要諮詢外部委員或提送藥品諮議小組討論。
- 若對函文內容有疑問,可連絡案件承辦人或申請審查案件函文釋疑。
- 有關案件審查進度,可至衛生福利部食品藥物管理署網站>藥品>案件申辦進度查詢。
- 配合藥政管理電子化,申請者於收到仿單核定草本後,應至線上平台進行仿單建檔或變更作業。
- 諮詢服務
本中心具豐富的國內外法規科學知識及多年審查經驗,提供業者及研發單位在藥品於研發至上市各階段多元法規諮詢服務,歡迎各界先進多加利用,以利準備符合法規要求之送審文件,如欲提出申請,請詳見諮詢服務。
請依據衛生福利部公布最新之「西藥查驗登記審查費收費標準」繳納規費。